Presentada la primera guia clínica sobre la síndrome de Schaaf-Yang per a professionals i famílies
Millorar el coneixement de la síndrome de Schaaf-Yang (SYS) -una malaltia ultrarara causada per mutacions en el gen MAGEL2- és l'objectiu de la primera guia clínica dirigida a professionals sanitaris i famílies d'infants afectats per aquesta patologia.
El treball, publicat a la revista Journal of Medical Genetics, ha permès conèixer quins efectes té la proteïna truncada de MAGEL2 en la fisiologia de les cèl·lules.
L'article es basa en un projecte liderat per les investigadores Roser Urreizti, investigadora del Centre d'Investigació Biomèdica en Xarxa de Malalties Rares adscrita a l'Institut de Recerca Sant Joan de Déu (IRSJD) i l'Hospital Sant Joan de Déu Barcelona, i Raquel Rabionet i Susanna Balcells, de l'IRSJD, de la Facultat de Biologia i de l'Institut de Biomedicina (IBUB) de la Universitat de Barcelona i del CIBERER, amb la participació d'equips investigadors de les institucions esmentades.
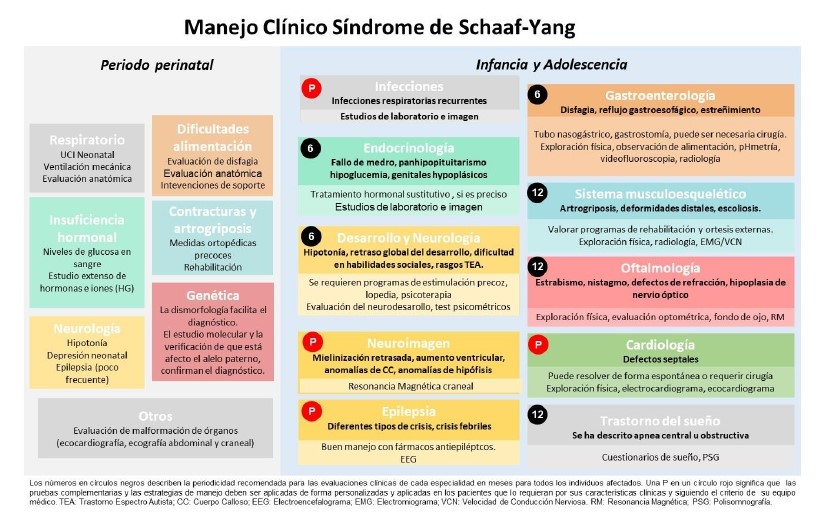
La síndrome de Schaaf-Yang (SYS) és una malaltia ultrarara causada per mutacions en el gen MAGEL2, localitzat al cromosoma 15. El gen MAGEL2 s'expressa principalment al cervell i la seva proteïna és clau en el complex de transport i la regulació del reciclatge de determinades proteïnes dins de les cèl·lules. Els pacients amb SYS presenten una àmplia varietat de símptomes i signes clínics, els més habituals de la malaltia són el retard del neurodesenvolupament i el retard intel·lectual, el trastorn de la son, la dismòrfia facial i les contractures articulars.
La implicació de les famílies i les associacions és clau en la recerca de les malalties rares
La soledat de tenir una malaltia ultrarara és una de les grans dificultats a les quals han de fer front les famílies afectades. Actualment, a Espanya només es coneixen 11 casos de síndrome de Schaaf-Yang, i menys de 200 arreu del món. Aquesta baixa incidència dificulta l'atenció mèdica estandarditzada, ja que no es disposa de pautes clíniques clares ni de tractaments o seguiments específics.
«Moltes vegades, les famílies i els professionals ens enfrontem a una gran incertesa davant la falta d'informació. Per aquest motiu, és vital comptar amb recomanacions basades en l'evidència que ajudin a millorar l'atenció clínica i a empoderar les famílies sobre el curs natural de la malaltia», comenta Merche Serrano, neuropediatra de l'Hospital Sant Joan de Déu Barcelona, investigadora de l'IRSJD i coordinadora de la part clínica de l'estudi.
Una guia per a professionals i famílies basada en l'evidència
L'equip de recerca va dur a terme una revisió de tots els articles i estudis que s'havien publicat sobre la SYS, amb l'objectiu d'elaborar una guia clínica extensa per a professionals mèdics i famílies, i que els dos col·lectius tinguessin més coneixements de la malaltia a l'hora de tenir cura dels pacients.
La guia clínica inclou els problemes mèdics més rellevants dividits en dos períodes de temps: un primer, que inclou els primers 28 dies de vida, i el segon, que comprèn la infància i l'adolescència. També s'han indicat les intervencions més adequades per a cada problema, a més de quines podrien ser les millors pautes de seguiment. Amb aquesta guia es vol garantir la millor atenció possible a tots els infants.
«També hem elaborat una guia i material dirigit a famílies, a petició de l'Associació de Famílies de la Síndrome de Schaaf-Yang, que els ajudarà a conèixer quina és l'evolució de la malaltia i a empoderar-los», diu Roser Urreizti. «Per a nosaltres, és molt important treballar de manera ben estreta amb les famílies i els pacients, sobretot en les malalties minoritàries en què els pacients tenen un paper clau per fer avançar la recerca», conclou la investigadora.
L'efecte tòxic de la proteïna truncada MAGEL2
L'extensa revisió de la literatura científica també ha permès elaborar un mapa de totes les mutacions del gen MAGEL2, la majoria de les quals produeixen una proteïna truncada (sense funcionalitat) o una proteïna amb una falta parcial o total del domini MHD, que deriva en un mal funcionament de la proteïna.
El gen MAGEL2 es troba al cromosoma 15 i produeix la proteïna MAGEL2, que forma part del complex de transport retrògrad i de reciclatge de proteïnes endosòmiques, a més de desenvolupar altres funcions.
«Els estudis que hem fet ens revelen que la proteïna MAGEL2 mutada queda retinguda al nucli de les cèl·lules, on podria alterar l'expressió i la regulació d'altres gens, mentre exerceix un efecte tòxic sobre la cèl·lula. En els fibroblasts dels pacients vam observar nivells baixos de beta amiloide (Aß) 1-40 i glutamina intracel·lular. Aquestes dues molècules ens podrien servir com a possibles biomarcadors per avaluar futurs tractaments», assegura Laura Castilla, primera autora del treball (UB-IBUB-IRSJD-CIBERER).
Els resultats confirmen que el fenotip de la síndrome de Schaaf-Yang el podria causar aquest efecte tòxic de la proteïna MAGEL2 truncada, més que no pas per la falta de funcionalitat normal, tal com es va observar en els fibroblasts dels pacients estudiats.
Les investigadores predoctorals Mònica Centeno i Aina Prat continuaran aquests estudis amb l'anàlisi de la funció de MAGEL2 en organoides de cervell generats a partir de cèl·lules pluripotents derivades dels pacients.
Article de referència
Castilla-Vallmanya, L.; Centeno-Pla, M.; Serrano, M. et al. «Advancing in Schaaf-Yang syndrome pathophysiology: from bedside to subcellular analyses of truncated MAGEL2». Journal of Medical Genetics, setembre de 2022. Doi:10.1136/ jmedgenet-2022-108690
Els estudis que hem fet ens revelen que la proteïna MAGEL2 mutada queda retinguda al nucli de les cèl·lules, on podria alterar l'expressió i la regulació d'altres gens, mentre exerceix un efecte tòxic sobre la cèl·lula.